Hi there!
Early this year we published a research about getting animal abundance estimates from wildlife - vehicles collisions in Ecography journal:
Can we model distribution of population abundance from wildlife–vehicles collision data?

The first thing we had to deal with was to obtain geographical coordinates (latitude and longitude) from a database with the name of the roads and the kilometer point. At the end, I wrote a small chunk of code to do that by using the Transport Network from the Spanish Geographic Institute. I first downloaded all the kilometer point datasets (by provinces) and I merged them all in a single file. You can find this file here (but it's old and current datasets should be updated, so I recommend you to download it again from the original sources). Then I used R language to create a routine to "search" the coordinates that better matched my points.
I recently updated my code, warping it in a function and I created a toy-dataset with 5 points to show you how it works (see below, comments in Spanish).
library(dplyr)
library(RCurl)
# Guardamos las URLs de los datos almacenados en mi Github (https://github.com/jabiologo/)
roadData <- "https://github.com/jabiologo/roads/blob/main/roadData.RData?raw=true"
# Cargamos la base de datos del IGM con los puntos kilométricos
# Cargamos la base de datos con nuestros nombres de carreteras y kilómetros
load(url(roadData))
# Base de datos del IGN
head(km)
# Mis puntos para los cuales quier las coordenadas
# Nótese que si se quieren utilizar unos datos propios con este script,
# estos deben tener el mismo formato que el objeto "puntos"
head(puntos)

# Cargamos la función
pkm2xy <- function(events, database){
# Almacenaremos todas las carreteras que no se encuentren en nuestra base de datos
noMatch <- events[!(events[,1] %in% database$Nombre),]
# Filtramos nuestros datos
events <- events[events[,1] %in% database$Nombre,]
# Creamos una serie de columas para almacenar las coordenadas y otra info
events$X <- NA
events$Y <- NA
events$error <- NA
events$RoadAssigned <- NA
events$PkmAssigned <- NA
# Mediante un bucle iremos obteniendo nuestras coordenadas
for(i in 1:nrow(events)){
# Seleccionamos de nuestra base de datos aquel punto kilométrico que se
# encuentre más cerca de nuestro kilómetro
sel <- database %>% filter(Nombre == events[i,1]) %>%
filter(abs(numero - events[i,2]) == min(abs(numero - events[i,2])))
# Almacenamos las coordenadas
events[i,3:4] <- sel[1,1:2]
# Calculamos el error entre el punto kilométrico obtenido y el deseado
events[i,5] <- abs(sel$numero[1] - events[i,2])
# Almacenamos el nombre de la carretera y el punto kilométrico que hemos
# seleccionado en nuestra base de datos del IGN
events[i,6] <- sel$Nombre[1]
events[i,7] <- sel$numero[1]
print(i)
}
# Retornamos una lista con dos elementos:
# Aquellos puntos para los cuales se han conseguido las coordenadas
# Aquellos puntos para los que no se ha encontrado la carretera
return(list(events,noMatch))
}
# Corremos nuestra función con el fichero de ejemplo
pkm2xy(puntos,km)
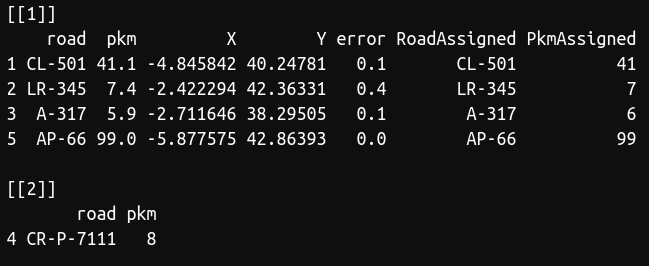
As you can see, there was a road (CR-P-7111) that was not present in our database, so we couldn't find the coordinates for this point. However, we obtained the coordinates for the rest points. The function also show you the error in kilometers between the point and the latitude/longitude obtained. I recommend to perform a random test to visually check that everything was right, that is, if your dataset is large, select a number of points and manually check with Google Maps or similar if the function worked well.
You can find the code and the data also in my GitHub repository.
Hope it's useful!
Best
References













